Local properties of individual magnetic sites
It is almost always very useful to analyze magnetic systems in terms of the individual magnetic sites of the crystal. In this tutorial, we illustrate how to calculate individual site properties for the magnetic atoms in GPAW.
Since it is not well-defined a priori where one site ends and another begins, GPAW supplies functionality to calculate the site properties as a function of spherical radii \(r_\mathrm{c}\). In this picture, the site properties are defined in terms of integrals with unit step functions \(\Theta(\mathbf{r}\in\Omega_{a})\), which are nonzero only inside a sphere of radius \(r_\mathrm{c}\) around the given magnetic atom \(a\).
Local functionals of the spin-density
For any functional of the (spin-)density \(f[n, \mathbf{m}](\mathbf{r})\), one may define a corresponding site quantity,
GPAW supplies functionality to compute such site quantities defined based on local functionals of the spin-density for collinear systems, \(f[n,\mathbf{m}](\mathbf{r}) = f(n(\mathbf{r}),n^z(\mathbf{r}))\). The implementation (using the PAW method) is documented in [1].
In particular, the site magnetization,
can be calculated via the function
calculate_site_magnetization(), whereas the function
calculate_site_zeeman_energy() computes the LSDA site
Zeeman energy,
Example: Iron
In the script
Fe_site_properties.py,
the site magnetization and Zeeman energy are calculated from the ground state
of bcc iron. The script should take less than 10 minutes on a 40 core node.
After running the calculation script, you can download and execute
Fe_plot_site_properties.py
to plot the site magnetization and Zeeman energy as a function of the
spherical site radius \(r_\mathrm{c}\).

Although there does not exist an a priori magnetic site radius \(r_\mathrm{c}\),
we clearly see that there is a region, where the site Zeeman energy is constant
as a function of the radius, hence making \(E_a^\mathrm{Z}\) a well-defined
property of the system in its own right.
However, the same cannot be said for the site magnetization, which continues to
vary as a function of the cutoff radius. This is due to the fact that the
interstitial region between the Fe atoms is slightly spin-polarized
anti-parallel to the local magnetic moments, resulting in a radius
\(r_\mathrm{c}^\mathrm{max}\) which maximizes the site magnetization (marked with
a dotted line). If one wants to employ a rigid spin approximation for the
magnetic site, i.e. to assume that the direction of magnetization is constant
within the site volume, it is natural to choose \(r_\mathrm{c}^\mathrm{max}\) to
define the sites. In practice, \(r_\mathrm{c}^\mathrm{max}\) can be calculated
(along with the maximized magnetic moment) via the function
maximize_site_magnetization() and in general, the
allowed ranges of atomic cutoff radii can be inspected via the
get_site_radii_range() function.
Site-based sum rules
In addition to site quantities, one may also introduce the concept of site matrix elements, that is, expectation values of functionals \(f(\mathbf{r})=f[n, \mathbf{m}](\mathbf{r})\) evaluated on specific spherical sites,
Similar to the site quantities, GPAW includes functionality to calculate site matrix elements for arbitrary local functionals of the (spin-)density \(f(\mathbf{r}) = f(n(\mathbf{r}),n^z(\mathbf{r}))\), with implementation details documented in [1]. For example, one can calculate the site pair density
as well as the site Zeeman pair energy
Now, from such site matrix elements, one can formulate a series of sum rules for various site quantities. For instance, one can construct single-particle sum rules for both the site magnetization and the site Zeeman energy, simply by summing over the diagonal of the site matrix elements for all the occupied states, weighted by the Pauli matrix \(\sigma^z\),
Although trivial, these sum rules can be used as a consistency tests for the
implementation and can be accessed via the functions
calculate_single_particle_site_magnetization() and
calculate_single_particle_site_zeeman_energy().
In addition to the single-particle sum rules, one may also introduce actual pair functions that characterize the band transitions of the system. In particular, one may introduce the so-called pair site magnetization
and pair site Zeeman energy
which turn out to be \(\mathbf{q}\)-independent diagonal pair functions,
\(m_{ab}(\mathbf{q})=\delta_{ab} m_a\) and
\(E^{\mathrm{Z}}_{ab}(\mathbf{q})=\delta_{ab} E^\mathrm{Z}_a\),
thanks to a simple sum rule [1]. Because the sum rule relies on the
completeness of the Kohn-Sham eigenstates, it breaks down when using only a
finite number of bands. Hence, it can be useful to study the band convergence of
\(m_{ab}(\mathbf{q})\) and \(E^{\mathrm{Z}}_{ab}(\mathbf{q})\) to gain insight
about related completeness issues of more complicated pair functions. In GPAW,
they can be calculated using the
calculate_pair_site_magnetization() and
calculate_pair_site_zeeman_energy() functions.
Example: Iron
In the
Fe_site_sum_rules.py
script, the single-particle site Zeeman energy is calculated along with the
pair site Zeeman energy using a varying number of bands. It should take less
than half an hour on a 40 core node to run.
Having done so, you can execute
Fe_plot_site_sum_rules.py
to plot the band convergence of \(E^{\mathrm{Z}}_{ab}(\mathbf{q})\).

Whereas the single-particle site Zeeman energy (dotted line) is virtually identical to the Zeeman energy calculated from the spin-density (blue line), there are significant deviations from the two-particle site Zeeman energy sum rule, especially with a low number of bands. Including at least 12 bands beyond the 4s and 3d valence bands, we obtain a reasonable fulfillment of the sum rule in the region of radii, where the site Zeeman energy is flat. Interestingly, this is not the case at smaller site radii, meaning that the remaining incompleteness shifts the site Zeeman energy away from the nucleus, while remaining approximately constant when integrating out the entire augmentation sphere.
In the figure, we have left out the imaginary part of the pair site Zeeman energy. You can check yourself that it vanishes more or less identically.
Exchange parameters
Although site-based sum rules can be enlightening in terms of PAW completeness and internal consistency of the code, the site matrix elements only bring real novelty when used for calculation of properties which can’t be obtained as simple functionals of the local (spin-)density. The Heisenberg exchange constants constitute exactly such physical quantities. In the rigid spin approximation, the lattice Fourier transformed exchange constants are given by (see e.g. [1])
where \(J(\mathbf{r}, \mathbf{r}', \mathbf{q})\) is the lattice Fourier transform of the exchange field \(J(\mathbf{r}, \mathbf{r}')\):
In the linear response formulation of the magnetic force theorem, the exchange field can by approximated within the LDA as [2]
where \(B^\mathrm{xc}(\mathbf{r})=-\left|W_\mathrm{xc}^z(\mathbf{r})\right|\) crucially is a local functional of the spin-density, see also Magnon dispersion from the magnetic force theorem. Consequently, the exchange constants can be written on the form of a site pair function
where the spin pair energy site matrix elements,
are intimately related to the site Zeeman pair energy. To calculate Heisenberg
exchange constants in this way, you can use the GPAW function
calculate_exchange_parameters().
If you do so, please reference both of the works [1] and [2].
Example: Cobalt
In the
Co_exchange_parameters.py
script, we calculate the Co exchange parameters \(\bar{J}^{ab}(\mathbf{q})\) as
a function of the spherical site cutoff radius \(r_\mathrm{c}\) for the \(\Gamma\),
M, K and A high-symmetry points. Furthermore we calculate exchange constants at
the ideal rigid spin approximation cutoff \(r_\mathrm{c}^\mathrm{max}\) for all
commensurate q-points along the corresponding \(\Gamma\)-M-K-\(\Gamma\)-A
high-symmetry path. It should take less than an hour on a 40 core node to run.
You can then execute
Co_plot_hsp_magnons_vs_rc.py
to examine the \(r_\mathrm{c}\)-dependence of the exchange parameters via the
resulting high-symmetry point magnon energies.

Once again, there exists a range of cutoff radii \(r_\mathrm{c}\) where key magnetic quantities are not sensitive to \(r_\mathrm{c}\)’s actual value, in this case the magnon energies calculated with the site magnetization held constant. Thus, despite the lack of an a priori definition for the extension of the Co magnetic sites, one may nevertheless take the isotropic exchange \(\bar{J}^{ab}(\mathbf{q})\) resulting from a projection of the exchange field onto atom-centered spherical sites to be a well-defined physical property of the hcp-Co system.
Executing
Co_plot_dispersion.py,
you can explore the full magnon dispersion of hcp-Co as calculated within LDA in
the LR-MFT method.

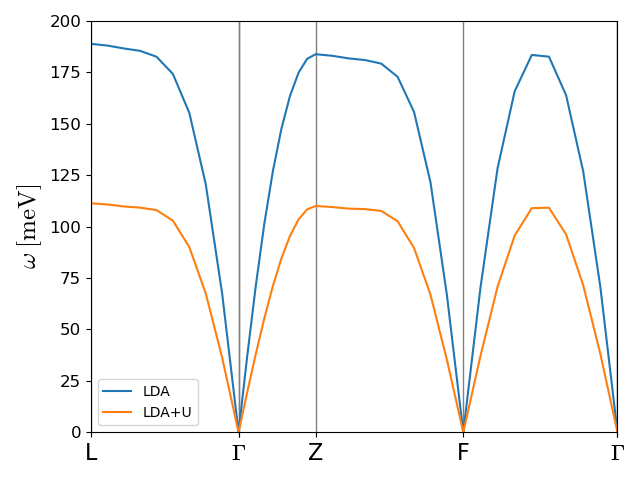
Example: NiO with LDA+U
For strongly correlated systems the exchange splitting is often underestimated, which results in too large exchange constants. This may largely be remedied by inclusion of Hubbard corrections through the LDA+U scheme. This approach introduces an additional term in the KS Hamiltonian that needs to be included in addition to the exchange-correlation magnetic field. This is easily included in the local site formulation and results in a U-dependent correction to the spin pair energy site matrix element defined above.
The effect of U is be illustrated by the anti-ferromagnet NiO here. The exchange
constants with and without Hubbard corrections can be calculated with the script
nio_dispersion.py, which takes roughly 10 hours on 120 CPUs. The
spin-wave dispersions with and without U is calculated and plotted with the script
plot_nio_dispersion.py and are show below. Experimentally, the magnon
band width is close to 110 meV, which is close to the LDA+U result.
Excercises
To get comfortable with the presented functionality, here are some suggested exercises to get you started:
Calculate the site pair magnetization of iron and analyze its band convergence.
Investigate the sensitivity of the site pair functions as a function of the wave vector \(\mathbf{q}\).
Calculate the site magnetization and Zeeman energy for a ferromagnetic material with inequivalent magnetic sublattices.
Are you still able to find ranges of radii, where the site Zeeman energy is constant?
What happens to the band convergence of the pair functions?
How does the off-diagonal elements of the pair functions converge as a function of the number of bands?
Calculate and plot the \(r_\mathrm{c}\)-dependence of the full magnon dispersion in hcp-Co. Does the conclusion that the LR-MFT magnon dispersion is a well-defined quantity hold also in this more general case?
Calculate and plot the band-convergence of the Co magnon energies. Is there a similar interplay between the number of bands and its \(r_\mathrm{c}\)-sensitivity as shown for the pair site Zeeman energy of Fe?
Calculate and plot the magnon band width (maximal magnins energy )as a function of U for the case of anti-ferromagnetic NiO.
API
- gpaw.response.site_data.calculate_site_magnetization(gs, sites)[source]
Calculate the site magnetization.
- Returns:
magmom_ap – Magnetic moment in μB of site a under partitioning p, calculated directly from the ground state density.
- Return type:
np.ndarray
- gpaw.response.site_data.calculate_site_zeeman_energy(gs, sites)[source]
Calculate the site Zeeman energy.
- Returns:
EZ_ap – Local Zeeman energy in eV of site a under partitioning p, calculated directly from the ground state density.
- Return type:
np.ndarray
- gpaw.response.site_data.maximize_site_magnetization(gs, indices=None)[source]
Find the allowed site radii which maximize the site magnetization.
Assumes that m(rc) is maximized for some rc belonging to the interior of the allowed cutoff radii for each atom. Physically, m(rc) has such a maximum only if the spin-polarization of the interstitial region is anti-parallel to the site in its near vicinity.
- Returns:
rmax_a (np.ndarray) – Cutoff radius in Å, maximizing the site magnetization for each site a.
mmax_a (np.ndarray) – Site magnetization in μB at its maximum for each site a.
- gpaw.response.site_data.get_site_radii_range(gs)[source]
Get the range of valid site radii for the atoms of a given ground state.
- Returns:
rmin_A (np.ndarray) – Minimum cutoff radius in Å for each atom A.
rmax_A (np.ndarray) – Maximum cutoff radius in Å for each atom A.
- gpaw.response.mft.calculate_single_particle_site_magnetization(gs, sites, context='-')[source]
Calculate the single-particle site magnetization.
- Returns:
sp_magmom_ap – Magnetic moment in μB of site a under partitioning p, calculated based on a single-particle sum rule.
- Return type:
np.ndarray
- gpaw.response.mft.calculate_single_particle_site_zeeman_energy(gs, sites, context='-')[source]
Calculate the single-particle site Zeeman energy.
- Returns:
sp_EZ_ap – Local Zeeman energy in eV of site a under partitioning p, calculated based on a single-particle sum rule.
- Return type:
np.ndarray
- gpaw.response.mft.calculate_pair_site_magnetization(gs, sites, context='-', q_c=[0.0, 0.0, 0.0], nbands=None, nblocks=1)[source]
Calculate the pair site magnetization.
- Parameters:
q_c (Vector) – q-vector to evaluate the pair site magnetization for.
nbands (int or None) – Number of bands to include in the band summation of the pair site magnetization. If nbands is None, it includes all bands.
nblocks (int or str) – The workload is parallelized over k-points and band+spin transitions. The latter is divided into nblocks, integrating nprocessors / nblocks k-points at a time.
- Returns:
magmom_abp – Pair magnetization in μB of site a and b under partitioning p, calculated based on a two-particle sum rule.
- Return type:
np.ndarray
- gpaw.response.mft.calculate_pair_site_zeeman_energy(gs, sites, context='-', q_c=[0.0, 0.0, 0.0], nbands=None, nblocks=1)[source]
Calculate the pair site Zeeman energy.
- Parameters:
q_c (Vector) – q-vector to evaluate the pair site Zeeman energy for.
nbands (int or None) – Number of bands to include in the band summation of the pair site Zeeman energy. If nbands is None, it includes all bands.
nblocks (int or str) – The workload is parallelized over k-points and band+spin transitions. The latter is divided into nblocks, integrating nprocessors / nblocks k-points at a time.
- Returns:
EZ_abp – Local pair Zeeman energy in eV of site a and b under partitioning p, calculated based on a two-particle sum rule.
- Return type:
np.ndarray
- gpaw.response.mft.calculate_exchange_parameters(gs, sites, q_c, context='-', nbands=None, nblocks=1)[source]
Calculate the Heisenberg exchange parameters.
- Parameters:
q_c (Vector) – q-vector to evaluate the pair site Zeeman energy for.
nbands (int or None) – Number of bands to include in the band summation of the pair site Zeeman energy. If nbands is None, it includes all bands.
nblocks (int or str) – The workload is parallelized over k-points and band+spin transitions. The latter is divided into nblocks, integrating nprocessors / nblocks k-points at a time.
- Returns:
J_abp – Heisenberg exchange parameters in eV of sites a and b under partitioning p.
- Return type:
np.ndarray